Amber Biology principal is co-author on paper on the evolution of transcriptional networks
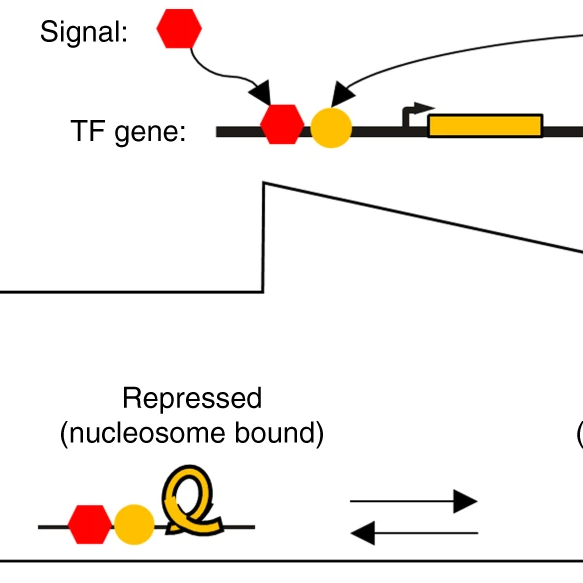
Here at Amber Biology we build computational models of a wide-range of cellular systems, from transcription networks, through to metabolic and cell-signaling pathways. Mechanistic computational models, particularly rule-based stochastic models, are a vital complement to wet-lab experiments. Computational models can also provide insights into evolution and indeed a paper just published in Nature Communications co-authored by Amber Biology Principal Alex Lancaster, asks whether the 3-node feed-forward loop motif, widely hypothesized to have evolved to filter out spurious signals, actually evolved for that purpose. Due to it’s overrepresentation in the transcriptional networks of many species, and it’s demonstrated function as a filter, many researchers have previously accepted this ‘just-so’ account of the feed-forward motif's adaptive origin. To test the adaptive hypothesis properly, the team built a detailed stochastic model of the dynamics of transcriptional networks, and then allowed the network to evolve under selection for the filtering function, and without selection for the function to see under which scenarios the motif appears (see also last year’s Ronin Institute seminar).
Spoiler alert: it looks like selection was responsible, but we learned much much more, including the fact that different kinds of motifs also can evolve for the same filtering function. The study reinforces the important role that in-silico approaches have in understanding the noisy world of biological systems. The paper is open-access and you can read the work in full in the link below, all code is also available on GitHub.
Read full open-access paper Xiong, Lancaster, Siegal, Masel (2019) at Nature Communications ...